
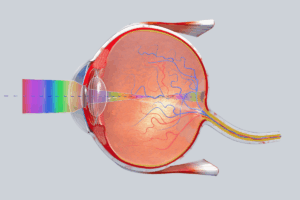
World’s First Human Implant of a 3D-Printed Cornea Restores Sight
December 15, 2025




Background
Epidemiology
Anatomy
Pathophysiology
The pathophysiology of CDKL5 Deficiency Disorder is linked to mutations in the cyclin-dependent kinase-like 5 (CDKL5) gene located on the X chromosome. The CDKL5 gene provides instructions for producing the CDKL5 protein, which plays a crucial role in brain development and function.
The CDKL5 protein is part of a family of enzymes called kinases that regulate the activity of other proteins in cells. It is essential for the development and maturation of neurons in the brain and the formation and stability of synapses, which are the connections between neurons.
Mutations in the CDKL5 gene lead to a loss or dysfunction of the CDKL5 protein, which disrupts normal brain development and function. The exact mechanisms by which CDKL5 mutations cause the characteristic features of the disorder are not fully understood, but several hypotheses have been proposed:
Etiology
Genetics
Prognostic Factors
CDKL5 Deficiency Disorder is a complex condition with significant variability in symptoms and outcomes among affected individuals. CDKL5 Deficiency Disorder has a wide variety of clinical manifestations, making it difficult to predict long-term prognosis. However, certain prognostic factors have been identified that may provide some insight into the potential outcomes for individuals with this disorder:
Clinical History
CDKL5 Deficiency Disorder is a rare genetic disorder that primarily affects females. The clinical presentation of CDKL5 Deficiency Disorder can vary widely depending on the severity of the genetic mutation, and the age of onset can range from early infancy to later childhood.
The following are the clinical presentations of CDKL5 Deficiency Disorder in different age groups:
Associated comorbidities or activities that may affect the clinical presentation of CDKL5 Deficiency Disorder include:
The acuity of presentation of CDKL5 Deficiency Disorder can vary, with some infants presenting with severe developmental delays and seizures from birth. In contrast, others may have milder symptoms that become more apparent as they age. It is essential to monitor and evaluate individuals with CDKL5 Deficiency Disorder regularly to identify and manage associated medical issues and provide appropriate interventions and support.
Physical Examination
Physical examination findings in individuals with CDKL5 Deficiency Disorder can vary, but certain features may be observed. It is important to note that the clinical presentation can differ among individuals, and not all features may be present in every case. Here are some physical examination findings commonly associated with CDKL5 Deficiency Disorder:
Age group
Associated comorbidity
Associated activity
Acuity of presentation
Differential Diagnoses
CDKL5 Deficiency Disorder can share clinical features with other genetic and neurodevelopmental disorders. The following is a list of some conditions that may be considered in the differential diagnosis of CDKL5 Deficiency Disorder:
Laboratory Studies
Imaging Studies
Procedures
Histologic Findings
Staging
Treatment Paradigm
by Stage
by Modality
Chemotherapy
Radiation Therapy
Surgical Interventions
Hormone Therapy
Immunotherapy
Hyperthermia
Photodynamic Therapy
Stem Cell Transplant
Targeted Therapy
Palliative Care
Medication
Future Trends
References
The pathophysiology of CDKL5 Deficiency Disorder is linked to mutations in the cyclin-dependent kinase-like 5 (CDKL5) gene located on the X chromosome. The CDKL5 gene provides instructions for producing the CDKL5 protein, which plays a crucial role in brain development and function.
The CDKL5 protein is part of a family of enzymes called kinases that regulate the activity of other proteins in cells. It is essential for the development and maturation of neurons in the brain and the formation and stability of synapses, which are the connections between neurons.
Mutations in the CDKL5 gene lead to a loss or dysfunction of the CDKL5 protein, which disrupts normal brain development and function. The exact mechanisms by which CDKL5 mutations cause the characteristic features of the disorder are not fully understood, but several hypotheses have been proposed:
CDKL5 Deficiency Disorder is a complex condition with significant variability in symptoms and outcomes among affected individuals. CDKL5 Deficiency Disorder has a wide variety of clinical manifestations, making it difficult to predict long-term prognosis. However, certain prognostic factors have been identified that may provide some insight into the potential outcomes for individuals with this disorder:
CDKL5 Deficiency Disorder is a rare genetic disorder that primarily affects females. The clinical presentation of CDKL5 Deficiency Disorder can vary widely depending on the severity of the genetic mutation, and the age of onset can range from early infancy to later childhood.
The following are the clinical presentations of CDKL5 Deficiency Disorder in different age groups:
Associated comorbidities or activities that may affect the clinical presentation of CDKL5 Deficiency Disorder include:
The acuity of presentation of CDKL5 Deficiency Disorder can vary, with some infants presenting with severe developmental delays and seizures from birth. In contrast, others may have milder symptoms that become more apparent as they age. It is essential to monitor and evaluate individuals with CDKL5 Deficiency Disorder regularly to identify and manage associated medical issues and provide appropriate interventions and support.
Physical examination findings in individuals with CDKL5 Deficiency Disorder can vary, but certain features may be observed. It is important to note that the clinical presentation can differ among individuals, and not all features may be present in every case. Here are some physical examination findings commonly associated with CDKL5 Deficiency Disorder:
CDKL5 Deficiency Disorder can share clinical features with other genetic and neurodevelopmental disorders. The following is a list of some conditions that may be considered in the differential diagnosis of CDKL5 Deficiency Disorder:

The pathophysiology of CDKL5 Deficiency Disorder is linked to mutations in the cyclin-dependent kinase-like 5 (CDKL5) gene located on the X chromosome. The CDKL5 gene provides instructions for producing the CDKL5 protein, which plays a crucial role in brain development and function.
The CDKL5 protein is part of a family of enzymes called kinases that regulate the activity of other proteins in cells. It is essential for the development and maturation of neurons in the brain and the formation and stability of synapses, which are the connections between neurons.
Mutations in the CDKL5 gene lead to a loss or dysfunction of the CDKL5 protein, which disrupts normal brain development and function. The exact mechanisms by which CDKL5 mutations cause the characteristic features of the disorder are not fully understood, but several hypotheses have been proposed:
CDKL5 Deficiency Disorder is a complex condition with significant variability in symptoms and outcomes among affected individuals. CDKL5 Deficiency Disorder has a wide variety of clinical manifestations, making it difficult to predict long-term prognosis. However, certain prognostic factors have been identified that may provide some insight into the potential outcomes for individuals with this disorder:
CDKL5 Deficiency Disorder is a rare genetic disorder that primarily affects females. The clinical presentation of CDKL5 Deficiency Disorder can vary widely depending on the severity of the genetic mutation, and the age of onset can range from early infancy to later childhood.
The following are the clinical presentations of CDKL5 Deficiency Disorder in different age groups:
Associated comorbidities or activities that may affect the clinical presentation of CDKL5 Deficiency Disorder include:
The acuity of presentation of CDKL5 Deficiency Disorder can vary, with some infants presenting with severe developmental delays and seizures from birth. In contrast, others may have milder symptoms that become more apparent as they age. It is essential to monitor and evaluate individuals with CDKL5 Deficiency Disorder regularly to identify and manage associated medical issues and provide appropriate interventions and support.
Physical examination findings in individuals with CDKL5 Deficiency Disorder can vary, but certain features may be observed. It is important to note that the clinical presentation can differ among individuals, and not all features may be present in every case. Here are some physical examination findings commonly associated with CDKL5 Deficiency Disorder:
CDKL5 Deficiency Disorder can share clinical features with other genetic and neurodevelopmental disorders. The following is a list of some conditions that may be considered in the differential diagnosis of CDKL5 Deficiency Disorder:

Both our subscription plans include Free CME/CPD AMA PRA Category 1 credits.

On course completion, you will receive a full-sized presentation quality digital certificate.
A dynamic medical simulation platform designed to train healthcare professionals and students to effectively run code situations through an immersive hands-on experience in a live, interactive 3D environment.

When you have your licenses, certificates and CMEs in one place, it's easier to track your career growth. You can easily share these with hospitals as well, using your medtigo app.






