Medico-Legal Challenges in End-of-Life Interventions: Causes and Insights
April 8, 2026




Brand Name :
Tepmetko
Synonyms :
tepotinib
Class :
MET Tyrosine Kinase Inhibitors

Brand Name :
Tepmetko
Synonyms :
tepotinib
Class :
MET Tyrosine Kinase Inhibitors
Dosage forms and strengths
tablet
225mg
Safety and efficacy are not well established
Refer adult dosing
nafcillin will decrease the effect of action of tepotinib by affecting enzyme CYP3A4 metabolism.
the level of impact of tepotinib will increase by MDR1 P-glycoprotein efflux transporter.
CYP3A strong enhancers of the small intestine may reduce the bioavailability of tepotinib
increases serum level of idarubicin by P-glycoprotein efflux transporter
it increases the level of rifaximin with the help of the P-glycoprotein (MDR1) efflux transporter
it increases the effect of sitagliptin via the MDR1 efflux transporter
it increases the effect of linagliptin via the MDR1 efflux transporter
when both drugs are combined, there may be a reduced excretion rate of topotecan and result in an elevated level of serum concentration
Actions and Spectrum
The main action of drug is to selectively bind to the MET receptor, preventing its activation by its ligand, hepatocyte growth factor (HGF). By inhibiting MET activation, drug blocks downstream signaling pathways involved in tumor cell proliferation, invasion, and angiogenesis.
Its spectrum of activity is mainly focused on tumors with MET alterations, offering a targeted therapeutic option for patients with these specific genetic abnormalities.
Frequency defined
>10%
All grades
Decreased hemoglobin (27%)
Increased ALT (44%)
Decreased lymphocytes (48%)
Increased AST (35%)
Edema (70%)
Nausea (27%)
Decreased sodium (31%)
Increased alkaline phosphatase (50%)
Fatigue (27%)
Decreased albumin (76%)
Increased potassium (25%)
Diarrhea (26%)
Increased gamma-glutamyltransferase (24%)
Musculoskeletal pain (24%)
Increased creatinine (55%)
Grade 3 to 4
Decreased lymphocytes (11%)
1-10%
All grades
Headache (<10%)
Fever (<10%)
Pruritus (<10%)
Dizziness (<10%)
Rash (<10%)
ILD/pneumonitis (<10%)
Grade 3 to 4
Increased alkaline phosphatase (1.6%)
Increased gamma-glutamyltransferase (5%)
Decreased hemoglobin (2%)
Increased amylase (4.6%)
Pneumonia (3.9%)
Decreased sodium (8%)
Dyspnea (2%)
Increased ALT (4.1%)
Musculoskeletal pain (2.4%)
Increased AST (2.5%)
Decreased albumin (9%)
Pleural effusion (5%)
<1%
Grade 3 to 4
Cough (0.4%)
Abdominal pain (0.8%)
Increased creatinine (0.4%)
Diarrhea (0.4%)
Decreased leukocytes (0.8%)
Nausea (0.8%)
Black Box Warning:
None
Contraindication/Caution:
Hypersensitivity or allergic reaction:
If a patient has had an allergic reaction or hypersensitivity to tepotinib in the past, it should not be used again.
Pregnancy warnings:
Pregnancy category: AU TGA pregnancy category D
US FDA pregnancy category Not Assigned
Lactation: Excreted into human milk is unknown
Pregnancy Categories:
Pharmacology:
The drug is a small molecule inhibitor of the hepatocyte growth factor (HGF) receptor, commonly referred to as c-MET.
Pharmacodynamics:
The drug selectively inhibits the activation of the c-MET receptor, which plays a crucial role in cell growth, survival, and migration. Activation of c-MET signaling can lead to uncontrolled cell proliferation and metastasis in cancer. The drug binds to the ATP-binding site of c-MET and prevents its phosphorylation, thereby inhibiting downstream signaling pathways involved in cancer progression.
Pharmacokinetics:
Absorption
The drug is orally administered as a tablet. It is rapidly absorbed from the gastrointestinal tract, with peak plasma concentrations reached within approximately 2 hours after oral administration.
Distribution
The drug has a moderate volume of distribution, indicating that it is distributed throughout the body tissues. It is highly bound to plasma proteins (approximately 99.9%), primarily to albumin.
Metabolism
The metabolism of the drug take place in the liver through the cytochrome P450 enzyme system, particularly CYP3A4 and CYP2C9. The major circulating metabolite is M5, which is formed by oxidative metabolism of tepotinib.
Excretion and elimination
The drug and its metabolites are primarily eliminated via the feces (approximately 67%), while renal excretion accounts for a minor fraction (approximately 9%) of the total dose. The elimination half-life of tepotinib is approximately 40 hours.
Administration:
Dosage: The recommended dosage may vary depending on the individual’s condition. It is essential to follow the prescribed dose exactly as instructed by the healthcare provider.
Timing: The drug is usually taken once a day, with or without food. The specific time of day for administration may be recommended by the prescribing physician.
Patient information leaflet
Generic Name: tepotinib
Why do we use tepotinib?
Treatment of NSCLC with MET exon 14 skipping mutations: The drug has been approved for the treatment of NSCLC patients with MET exon 14 skipping mutations who have progressed on or are not eligible for platinum-based chemotherapy.
MET-driven cancers: The drug may also have potential in the treatment of other cancers that are driven by aberrant activation of the MET receptor, such as gastric cancer, hepatocellular carcinoma, and breast cancer.
Combination therapy: The drug is being studied in combination with other targeted therapies or immunotherapies to enhance the treatment outcomes in MET-driven cancers. For example, it is being investigated in combination with anti-PD-1/PD-L1 antibodies in NSCLC patients.
Research tool: The drug can be used as a research tool in preclinical and clinical studies to understand the biology of MET-driven cancers and to evaluate the effectiveness of targeting the MET pathway.

Both our subscription plans include Free CME/CPD AMA PRA Category 1 credits.

On course completion, you will receive a full-sized presentation quality digital certificate.
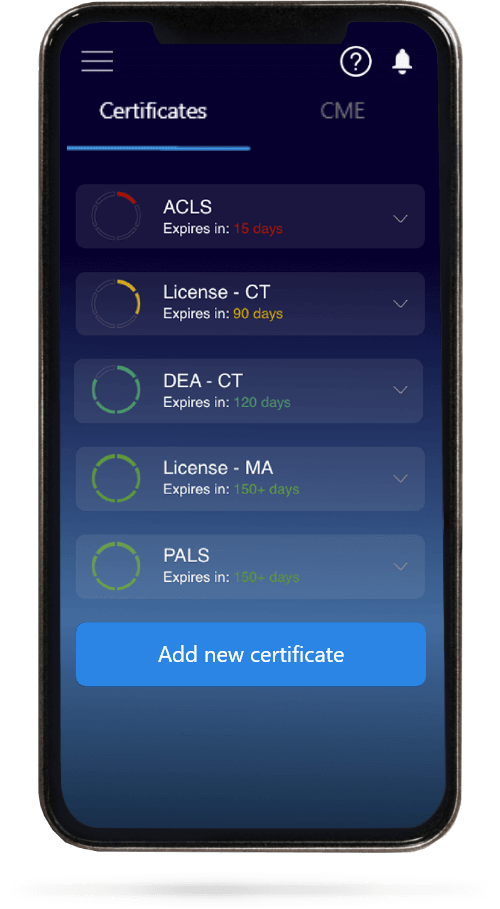
When you have your licenses, certificates and CMEs in one place, it's easier to track your career growth. You can easily share these with hospitals as well, using your medtigo app.